and genetic approaches
Go to: dictionary | proteins | cDNAs | antibodies | logic
& exptl design
(home page) | bioinformatics | GPIN
home Page
Forward. This section focuses on ways to bridge the gap between
|
|
|
|
This discussion focuses on vertebrate systems, but the logical principles are the same for all systems
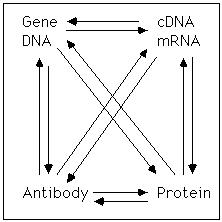
 The genetic information responsible for the synthesis
of messenger RNAs and generation of proteins resides in the genetic
material, which is usually DNA. Being able to understand and
manipulate genes is a powerful way to understand the function of RNA
and protein.
The genetic information responsible for the synthesis
of messenger RNAs and generation of proteins resides in the genetic
material, which is usually DNA. Being able to understand and
manipulate genes is a powerful way to understand the function of RNA
and protein.
Likewise, the field of genetics existed long before it was demonstrated that DNA was the genetic material, and from the point of view of a geneticist it is possible to understand a substantial amount about biology from the study of genes without even considering their physical nature. Likewise, the elucidation of the patterns of mRNA and protein synthesis can be seen as tools to get at the structure of genes and to understand the role and cellular processes and the development of an organism.
Why is it important to understand the molecular structure of genes? The answer to this question flows naturally considering what elements are present in the DNA that are not transcribed into mRNA.
All of these are interesting questions, but this section will focus on three questions:
How are genomic clones isolated? There are potential two routes to isolating a gene or a fragment of a gene, one beginning with a cDNA and the other beginning with a genetic trait and information about linkage.
Promoter analysis-the goals. The objective of promoter analysis is to understand what cis-acting DNA sequences are responsible for the regulation of gene expression and to understand how these sequences allow appropriate gene expression.
Defining the structure of cis-acting sequences by a functional assay. There is no a priori method of establishing what sequences are responsible for regulation of the expression of any gene. Essentially, the initial experiments must be based on a guess by the experimentalist of which sequences are likely to be important for regulation. These guesses can then be tested. If the test is correct, the guess can be refined to determine exactly what sequences are important for gene regulation. If it is incorrect the experimentalist must make another guess and test those hypotheses.
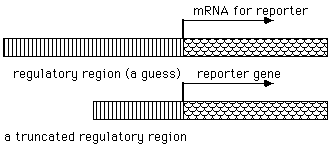 To test the assertion that a particular DNA sequence is
involved in the regulation of gene expression, it is necessary to
introduce those putative regulatory sequences into a cell and then
determine their activity. This is done by combining regulatory
sequence with an "reporter*" sequence that can be used to monitor the effect of
the regulatory sequences.
To test the assertion that a particular DNA sequence is
involved in the regulation of gene expression, it is necessary to
introduce those putative regulatory sequences into a cell and then
determine their activity. This is done by combining regulatory
sequence with an "reporter*" sequence that can be used to monitor the effect of
the regulatory sequences.
Reporter genes.* In general, reporter genes are chosen to be genes whose expression can be conveniently monitored. That is, the expression should be easily measured, there should be a minimum background, and there should be little interference from other genes that might be expressed by the cell. Currently, the most common reporter genes that are used are luciferase* and chloramphenicol acetyltransferase* (abbreviated CAT, and not to be confused with choline acetyl transferase, the gene that is responsible for the synthesis of the neurotransmitter acetylcholine and which is abbreviated CAT or ChAT). Luciferase is a gene originally isolated from the fire fly that in the presence of luciferin and ATP emits photon and production of photons can easily be monitored by a scintillation counter especially designed for this purpose. Chloramphenicol acetyltransferase is chosen because it is a bacterial gene that is not expressed in vertebrate cells. It too can be monitored because it can acetylate chloramphenicol and the acetylated chloramphenicol can be separated from unacetylated chloramphenicol by TLC and detected by the presence of label present in chloramphenicol. There is essentially no background level of activity in eucaryotic cells with this assay so it can be extremely sensitive. Both of these reporters have the advantage that they depend on the activity of a protein which is translated from a mRNA and so the translational process amplifies the signal.
It is also possible to measure RNA transcription directly by using an assay that uses either RNase protection* or northern analysis* to monitor mRNA levels. In some cases where the regulatory elements lie within the coding regions of the gene being studied it is often necessary to use a large part of the coding sequence of the gene to study transcriptional regulation. In these cases, introducing some kind of a marker into the reporter gene that allows it be distinguished from the endogenous gene can allow measurement of the transcriptional activity. For example it is possible to use a copy of an endogenous coding region that is modified by the addition or deletion of a restriction fragment or the incorporation of a novel restriction site. This strategy has the advantage that it is frequently possible to simultaneously measure the endogenous gene and the reporter gene which gives an additional control in the study of regulated transcriptional events.
Defining the limits of cis-acting sequences. Once a region containing a cis-acting* DNA sequence is identified the next challenge is to determine which specific sequences in the DNA are responsible for transcriptional activation or transcriptional repression. This is done by two strategies. Usually, it is most convenient to do deletion analysis first. That is deletions from the 5' and/or the 3' end of the regulatory region can be made and the shortest region of DNA that includes the regulatory effects can be determined. This approach can frequently shorten the area of interest from many thousands of bases to a few hundreds of bases.
More precise localization of DNA regulatory regions depends on site-directed mutagenesis. There are a number of approaches that allow the modification of any combination of bases in a DNA region. Such mutagenesis can provide powerful evidence for the exact binding sites of putative transcription factors.
Stable versus transient transfection analysis. The initial discussion of promoter analysis given above simply assumed that it was possible to introduce a reporter construct into a cell and measure the level of expression of a reporter under various conditions. In practice, there are 3 experimental difficulties that must be considered in executing any experiment of this type:
Stable transfection. Stable transfection refers to the production of a population of cells in which the gene being studied is stably expressed in the cell. Generally, this is thought to mean that the gene not only introduced into the cell but also integrated into the host DNA and carried along with it during cycles of cell division. In contrast, the initial plasmid that is introduced into a cell is generally thought to be episomal, which explains why it can frequently be lost or degraded. Studies of expression from a plasmid at this time are said to be transient because the DNA is only transiently present in most cells (see below for discussion of measuring expression at this time). To isolate the cells that are stably expressing a reporter construct it is necessary to eliminate cells that have failed to stably integrate that DNA of interest. This is done by transfecting cells, not only with the reporter construct of interest, but also with another plasmid carrying a selectable marker. Most frequently, this selectable marker is a gene for resistance to neomycin (G418). When cells are cotransfected with 2 plasmids, in the vast majority of cases (but not all cases) cells will integrate either both of these plasmids (and indeed in general multiple copies of both plasmids will either be integrated) or no copies will be integrated. Thus, selection for resistance to G418 will yield a population of cells that are expressing the reporter construct of interest. It is then possible to divide the cell population and study gene expression under a variety of different conditions. If one is interested in the ability of particular promoter sequences to respond to a variety of ligands, this strategy is an effective way to do those experiments. This approach has the experimental advantage that once isolated the cells can be used in multiple experiments and experiments can be repeated with ease. It has the disadvantage that it is necessary to go through a selection and growth of a sub population of cells that can be time consuming taking from a week to even months.
One of the experimentally important considerations that must be kept in mind in using stably transfected cells is that the DNA is integrated into the host chromosome. Depending on the site of integration, the flanking sequences are very likely to have strong influences on the expression of the DNA of interest. These influences may either increase or decrease the expression of the gene of interest. Thus, if a single transfected cell is isolated and studied the experimentalist may be studying the site of integration rather than the promoter elements present in the plasmid. To eliminate this as a problem, it is essential to study not single isolates but rather populations of isolated cells or multiple isolates. By studying a population consisting of thousands of clones it is more likely that any experimental clone effect seen will be a result of sequencing in the reporter construct than in the site of integration.
Transient transfection. The second general approach to doing transfection analysis is to do transient analysis. In this experiment DNA is introduced into a cell population by transfection, but no stable cell lines are isolated. Rather, gene expression is studied shortly after the transfection procedure usually within the 24-72 hours. This approach has the advantage that the experiments can be done relatively rapidly and that the same preparation of DNA can be introduced into many different cell types. It has the substantial disadvantage that the transfection efficiency in different preparations may be radically different so it is necessary to control for this transfection efficiency* if reliable data is to be obtained. To control for differences in transfection efficiency again the approach is to transfect not with a single plasmid of interest, but rather to transfect with 2 plasmids. The second one is a plasmid that is used to correct for transfection efficiency. The second plasmid is designed to express a gene that is easily assayed and whose expression is constitutive (i.e., it will not change under various experimental conditions). Thus, the expression of 2 reporter genes can be assayed in the cell population and it is the ratio of these 2 activities that indicates the efficiency of expression of the reporter gene and the activity of the promoter being studied.
Transfection procedures. How can DNA be introduced into a cell ? The cell membrane is a barrier to any molecule and a large highly charged molecule like DNA would be expected to have little success at entering the cell, much less the nucleus. A number of ways of overcoming this permeability barrier are available, and each one of these works effectively with certain cell types, so no general procedure has been established. Commonly used methods include :
All of these mechanisms can work effectively, but each has the disadvantage that they damage the cell and an optimal procedure is designed by testing various possibilities and balancing transfection efficiency with cell death.
Identification of transcription factors. The ultimate goal of transcriptional analysis is to determine the nature of binding protein that interact with specific DNA regulatory elements and to understand the mechanism of transcription. Of course not all transcription factors bind DNA directly. Some bind to another transcription factor or to a DNA-protein complex. It is possible to develop evidence for the existence of specific DNA-binding proteins by a variety of approaches but the most commonly used are DNA footprint analysis, gel shift analysis (also called gel retardation analysis), and methylation interference, which are described below. A web site devoted to these topics is found in a course website at the U of Arizona.
Foot print analysis* . Foot printing depends on the interaction of specific DNA-binding proteins with DNA and interference with reactions that are used to generate a DNA sequencing ladder.
Although the basic idea of doing a footprint is straight forward executing one in practice is more complex because of the difficulty of non-specific binding reactions. DNA is a highly charged molecule and many proteins may bind non-specifically to it and the challenge is to develop conditions where only more specific and high affinity DNA-interactions are visualized. To prevent non-specific interactions, it is necessary to titrate the reaction mix with either DNA or some type of DNA-like polymer to interact with and remove proteins that have the potential of interacting with the DNA of interest with low affinity. It is also possible although experimentally difficult, to carry out DNA footprinting reactions in vivo, but this will not be discussed here.
Gel shift analysis*. Another important way of studying DNA-proteins is by gel shift analysis. Again, this type of analysis is based on monitoring specific interactions between an oligonucleotide and DNA.
To do a gel shift analysis, a short region of DNA (typically 15-25 base pairs) is chosen and labeled. When fractionated on a gel, such an oligonucleotide normally runs extremely fast. If the oligonucleotide is first mixed with an extract containing DNA-binding proteins, the oligonucleotide may perform a stable interaction with a protein. Electrophoresis under non-denaturing conditions will result in a co-migration of the labeled oligonucleotide and the protein of interest. This change in migration (called either shift or retardation) is diagnostic for the existence of a DNA-binding protein.
The presence of a protein that can interact with a strongly charged DNA molecule is not of course unexpected and the real question is whether the protein that has been identified is interacting specifically with the DNA sequence in question (i.e., is it a high affinity, biologically important interaction). This can be addressed by doing competition experiments. If an excess of unlabeled authentic oligonucleotide is added to the reaction mix it should be able to compete with the labeled oligonucleotide for binding to the protein which is present at limiting concentrations and lead to a reduction in signal. On the other hand, the addition of an unrelated oligonucleotide should not lead to such a competition. Indeed, a specific DNA-binding proteins should interact with DNA in a way that is very dependent on the presence of specific DNA-protein contacts. Thus introduction of only a few specific mutations into the oligonucleotide should result in an oligonucleotide that is not capable of competing with the authentic oligonucleotide.
In many cases it is possible to use this technique to further identify DNA-binding proteins by combining immunological analysis with a gel shift analysis. If an antibody that recognizes a particular DNA-binding protein is available, this antibody may either interfere with the binding of the protein to a DNA, resulting in the loss of a band or it may form a complex with the transcription factor which is associated with the oligonucleotide leading to a change in its migration of a gel and a shift at a different mobility. Both of these can be useful ways of identifying the presence of specific transcription factors in a complex. Another way to identify the size of a DNA binding protein is provided by UV Cross-linking*.
Methylation interference* is a related approach. If some DNA bases are modified by methylation in vitro, that methylation will interfere with the formation of a DNA-protein complexes that are formed in vitro. If one analyzes the methylation pattern of DNA found in DNA-protein complexes with the methylation pattern of DNA that can't form a complex, the differences demonstrate the importance of specific DNA bases.
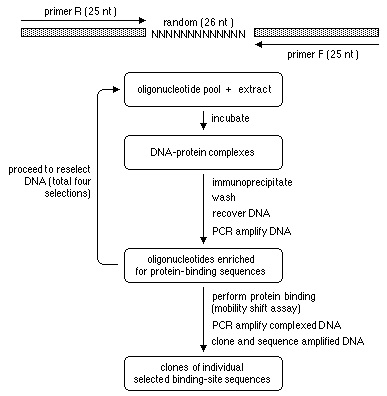
PCR-Assisted Binding Site Selection*. Another way to determine the binding site of a transcription factor (or another DNA binding protein) is to take advantage of its high affinity for a particular DNA sequence to select DNA containing that sequence from a collection (a library) of DNA sequences. To do this a library of random sequences is constructed with flanking primers so that it can be amplified. Affinity purification is used to enrich for the sequences that bind to the protein of interest, the selected sequences are amplified by PCR, and the procedure is repeated. A diagram of the procedure is available with its definition.
One hybrid approach to cloning transcription factors. If a cis acting sequence has been defined, it can sometimes be used to isolate the cDNA for the corresponding transcription factor on the basis of its ability to interact with the DNA sequence in yeast. Yeast containing appropriate reporter constructs are transfected with a library that contains fusion proteins between a cDNA library and a strong activator of transcription. Activation of the reporter means the clone is a candidate for the transcription factor of interest and additional criteria can test whether the clone is indeed the transcription factor of interest.
Bringing it
all together. At the beginning of
the section we indicated that the key idea of transcriptional
analysis was to show a relationship between the activity of
cis-acting DNA sequences and the transcription factors which they
associated. It is the combination of
---doing functional analysis of sequences and
---studying the biochemistry of transcription factors
which allows this to be done. If a particular transcription factor
responsible for a change in gene expression then changes in the
cis-acting DNA sequence that disrupt its binding should also result
in an inability to change transcriptional activity. By comparing the
physical and functional evidence for a particular DNA sequence it is
possible to make a persuasive case that a DNA-binding activity is
indeed a functional transcriptional factor. Yet again this is only
the first step in the analysis. Ultimately it is essential to purify
and clone the transcription factor. To understand how it actually
works it is necessary to reconstitute the enzymology of transcription
in vitro and understand interactions among transcription factors,
polymerases, and DNA elements.
From DNA sequence to genetic analysis (knock outs, knock ins, conditional knockouts, & trans genes)
Genetic systems. Different organisms provide different advantages (or disadvantages) for a genetic approach. In the case of Drosophila and yeast, it is possible to saturate a loci and produce a number of mutations including mutations that inactivate or disable a gene. It is possible to screen millions of organisms for an interesting phenotype. The ability to apply straight forward and powerful genetic technique is one of the things that makes some biological systems so experimentally tractable. For example, the ability to easily inactivate a gene by a process involving homologous recombination in yeast allows one to determine the phenotype of a mutation in any gene once a cDNA has been isolated.
On the other hand, the ability to apply genetics to vertebrate system has lagged behind. Homo Sapiens provides an incredible wealth of genetic information because the medical profession catalogs and categorizes interesting variations that might have a genetic basis and be amenable to genetic analysis. As the human genome project provides more and more markers on the human genome, this information will become more and more valuable. It is not easy to screen large number of vertebrates for interesting phenotype, although zebra fish are proving to be a promising experimental vertebrate system. There is no equivalent experimental system in mammalian species despite the fact mammalian species are of special interest to the biomedical scientist. Currently mice are the mammalian species best suited for genetics.
Because of this a variety of approaches have been developed that allow the production of mice with a defect in an identified gene using procedures that are based on homologous recombination. The only species where technology to do homologous recombination at will has been developed is the mouse; and, even, there the expense and commitment to make an animal defective in a known gene is substantial. On the other hand, techniques to introduce an additional gene into the germ line (a transgene*, see below) are available in many species, and this technology can be used to do genetic experiments which can either study the function of a protein by expression of the wild type protein or by expressing a mutant form of the protein. A mutant protein can have an effect on its own or it can exert an effect by interfering with the endogenous protein (by acting as a dominant negative).
In many cases, the expected phenotype of a particular mutant can be predicted (or guessed at), while in other cases the phenotype is completely unknown and the underlying question may be the general issue of whether an animal defective in a known loci will have a phenotype. In many cases it has turned out that there is no obvious phenotype in an animal carrying a complete deficiency in a gene product that was thought to be important (the predictions were completely wrong). In other cases the effect of the mutation is minimal. One caveat of such conclusions is always that finding a phenotype depends on the cleverness of the experimentalist and in some cases a phenotype may be subtle or only reveal itself under certain circumstances; nevertheless, a lack of an obvious phenotype is a clear signal that extensive study of that gene may be inadvisable.
How to make mice deficient in the product of a known gene. There are 2 problems that must be overcome in order to determine the effect of a mutation in a gene in the mouse.
Part one, making a mutation by homologous recombination*/gene targeting/knock-out* technology. The basic strategy used to disrupt a gene is to develop a targeting vector in which the sequence of a gene is interrupted in a coding region (exon) by a piece of DNA that will disrupt function. If such a "targeting vector" can recombine with the genomic loci by homologous recombination, the result will be an insertion into the gene of interest. The difficulty with such a simple strategy is that the frequency of homologous recombination in mice is extraordinarily low. In contrast, the frequency is high in yeast making this a relatively straight-forward procedure. To overcome this difficulty in mice, two strategies have been taken. First, the amount of homologous DNA in the targeting vector can be increased since recombination should be more frequent as the amount of homologous DNA is increased. Second, it is possible to incorporate 2 genetic selections into a vector.
Part two, getting the mutation into the germ line. To this point we have focused mainly on how it is possible to disrupt gene and such disruption can occur in any cell type in culture and this approach has been used extremely productively to determine the effect of a genetic mutation on tissue culture lines. However, the real power of this approach is that it is possible to create a mutagenic effect in certain cell lines which are subsequently capable of participating in embryogenesis and provide genetic material to a substantial part of a developing mouse. In this case, the cell chosen for insertional mutagenesis is a special cell type called embryonal carcinoma (EC) or an embryonic stem cell (ES)* . ES cells can be isolated and grown in culture as a continuous cell line and the manipulations is required for homologous recombination can be performed in these cells. The remarkable ability of these lines is that they can be selected and subsequently injected into a developing blastocyst. If this blastocyst, which has been isolated from a pregnant female, is subsequently re-injected into a pseudo-pregnant female, a mouse will develop in which some of the tissues are derived from the ES cells. Once the mice are born, this can be verified using a genetic marker. If the experimentalist is lucky enough that the ES cells contributed to the germ line of the mouse, the mouse can be bred and the mutation can be maintained. Using standard crossing techniques it is possible to bring the gene to homozygosity and test for biological function. The proceedures needed to get a knock out mouse are illustrated on another page
Making a mutant animal by genetic selection of mutant ES cells. One of the first mutant animals produced by this technology was made by doing a genetic selection against HGPRTase* in ES cells. The resulting mouse was HGPRTase deficient, but had no obvious phenotype. This was extremely disappointing because, in humans, the same deficiency causes mental retardation and a strong tendency to self mutilate by biting. It was hoped that the mouse could provide a model for this deficiency, but it didn't. The advantage of homologous recombination as an approach is that essentially any gene can be targeted and the gene can either be inactivated or modified at will.
Conditional knock outs. One of the problems with the approach described here is that many of the most interesting genes might be expected to have a lethal phenotype, so producing animals carrying such a mutation would only result in embryonic fatality and relatively little information. Likewise, when a more complex phenotype is studied (for example the ability to form memories or the function of a particular gene product in an adult organ system), the difficulty faced is that any changes seen may result, not from a change in the functioning of the gene product in the adult, but rather a change in the pattern of development. This is a frustrating logical conundrum that is not easy to address, but it led to a search for methods of developing methods of specifically inactivating a gene either in particular cell types or in particular developmental stages. These methods, which are based on the use of site-specific recombinases or the use of a regulated promoter are described below:
Site specific recombination (for a diagram see):
Conditional expression of a transgene. Another way of getting a conditional expression of a gene is to make a transgenic (see below) which uses a promoter whose expression is sensitive to an exogenous agent. A number of promoters may be suitable for this purpose, but two commonly used promoters include regulatory elements that are sensitive to tetracycline (an antibiotic) or ecdysone (a steroid hormone made by insects). Since there are no endogenous genes that respond to these compounds in mammalian cells, the presence of these promoters and the expression of tet-binding proteins or ecdysone binding proteins will have little effect on the function of endogenous genes. Generally, this strategy results in coordinate expression in all tissues, but more complex variations could restrict expression to unique tissue types. It is also possible to use this strategy to prevent the functioning of an endogenous gene by using the promoter to drive the expression of a dominant negative or to drive the expression of an antisense RNA.
Making more subtle mutations: the 'knock in'. Although it is often desirable to simply inactivate a gene to determine the importance of a null phenotype, in many cases it is more informative not to inactivate a gene, but rather to modify it so that its function is altered. Again, this is a task (which is the logical equivalent of site directed mutagenesis in a plasmid) that can be solved by homologous recombination. In this case a targeting vector is designed as a replacement vector so that additional genetic sequence are added into the genome. This technique is sometimes called a 'knock-in*'.
In situations where this has been effected, it is possible to subsequently select for loss of a selectable marker which would occur if there was inter chromosomal recombination leading to loss of genomic information. In some cases, the genomic information that is lost may be initially provided by the targeting vector, but it is equally possible that the genetic information is lost with the endogenous gene resulting in expression of the targeting vector which may have been designed to incorporate a more subtle mutation.
Producing organisms with an added gene product. It is also possible, and experimentally much easier, to produce an animal that expresses and additional gene, called a transgene. To do this, ES cells are transfected with an expression vector (promoter plus a coding sequence and a selectable marker). The transfected cells are then injected into a blastocyst and an animal can be produced by the same methods outlined above. This is much easier because there is no need to identify the rare cells where homologous recombination has occurred, but there are experimental difficulties that must be considered. The efficiency and tissue specificity of transgene expression will depend on the site of integration as well as the quality of the promoter chosen. Thus, transgenetics are exactly alike only if the gene is inserted into identical locations.
Conclusion. Thus, homologous recombination using targeting vectors that include both positive and negative selectable markers and incorporate either wild type or mutant sequences can be used to modify the genetic material in cell lines and in stem cells. If stem cells are used this genetic modification can be transmitted and the effect of a particular gene on the development of a whole organism can be determined. In some case it is possible to restrict the cell types where the genetic alteration occurs using a site specific recombinase. Thus, the power of genetic analysis can be brought to understanding the role of particular genes in a developing mammalian cell.
{kind=link}